
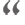 Polaryx Therapeutics, today announced that it has received from the U.S. Food and Drug Administration (FDA) both Rare Pediatric Disease and Orphan Drug Designations for the treatment of Niemann Pick Disease Types A and B, known as Acid Sphingomyelinase Deficiency with PLX-300.
Polaryx Therapeutics, today announced that it has received from the U.S. Food and Drug Administration (FDA) both Rare Pediatric Disease and Orphan Drug Designations for the treatment of Niemann Pick Disease Types A and B, known as Acid Sphingomyelinase Deficiency with PLX-300.




Polaryx Therapeutics, Inc., a biotech company developing small molecule therapeutics for lysosomal storage disorders, announced today that it has received from the U.S. Food and Drug Administration (FDA) both Rare Pediatric Disease and Orphan Drug Designations for the treatment of Niemann Pick Disease Types A and B, known as Acid Sphingomyelinase Deficiency with PLX-300.
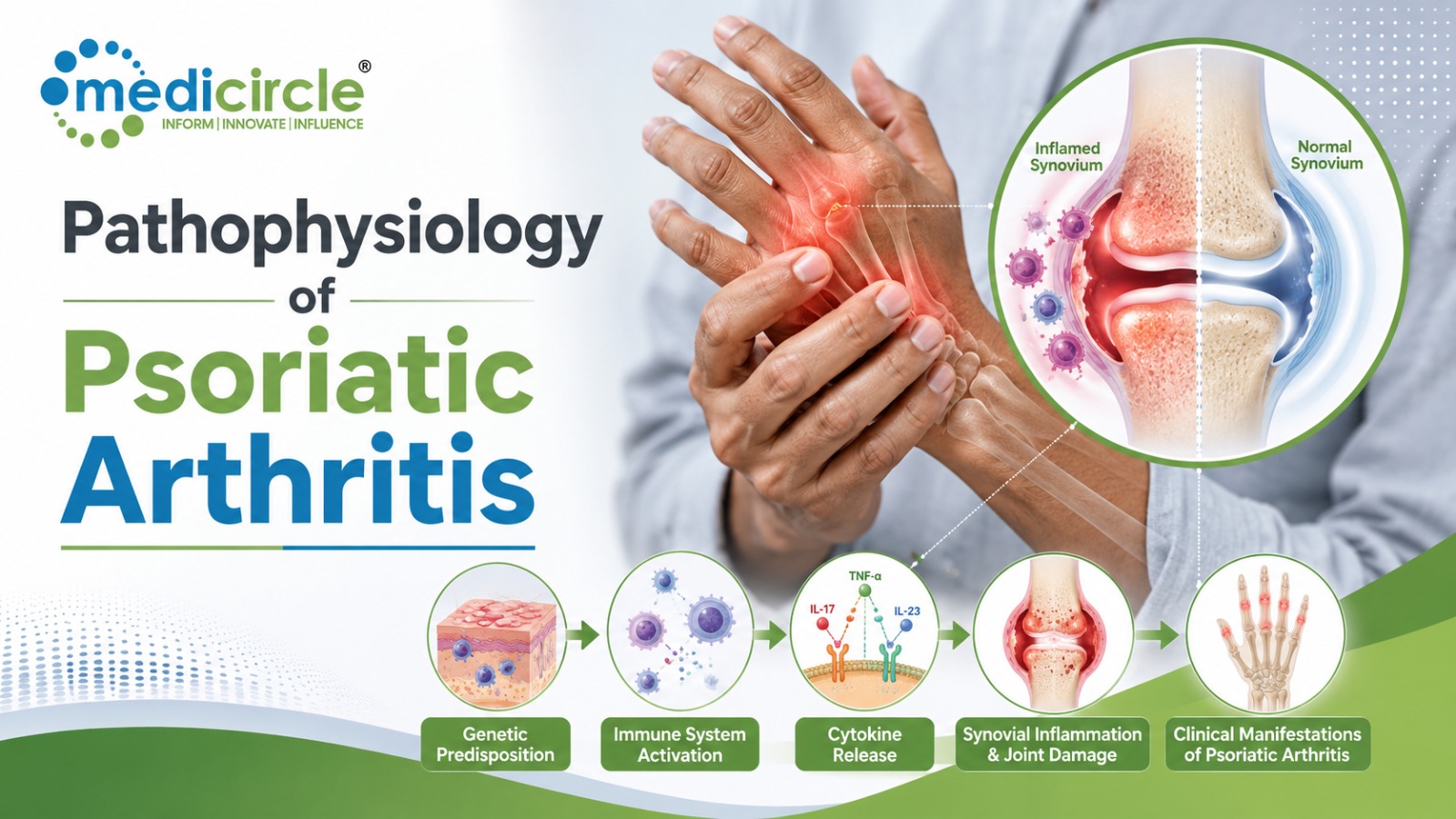
Niemann Pick Disease (NPD) Types A and B is a group of ultra-rare and fatal pediatric neurodegenerative disorders caused by defects in acid sphingomyelinase, a lysosomal enzyme that plays a pivotal role in the degradation of sphingomyelin. These genetic defects result in increased intracellular accumulation of sphingomyelin leading to severe neurodegeneration, cerebellar ataxia, and dementia. There is no cure for these diseases with the patients only receiving supportive care.
Under the FDA's rare pediatric disease designation program, the FDA grants Rare Pediatric Disease designation for serious or life-threatening diseases affecting fewer than 200,000 patients aged from birth to 18 years in the US. If a new drug application (NDA) for PLX-300 is approved, the Company is eligible to receive a priority review voucher that may be sold or transferred. In addition, because Orphan Drug Designation has been granted to PLX-300 for Niemann Pick Disease Types A and B from the FDA, the Company can receive the FDA's expedited review and approval process.
"Both Rare Pediatric Disease and Orphan Drug Designations to PLX-300 from the FDA for the treatment of Niemann Pick Disease Types A and B confirmed the scientific excellence of PLX-300 and its multi-indication potential to treat various lysosomal storage disorders. Recently, the FDA also has granted both Rare Pediatric Disease and Orphan Drug Designations to PLX-300 for GM2 gangliosidosis. We are now preparing IND-enabling studies to enter into Phase 1/2 studies as soon as possible" says Dr. Hahn-Jun Lee, M.Sc., Ph.D., President, and CEO of Polaryx Therapeutics, Inc.
Alex Yang, J.D., LLM, President and CEO of Mstone Partners Hong Kong and Chair of the Board at Polaryx Therapeutics, Inc., stated that "We are making tremendous steps towards developing several promising drug candidates to treat rare diseases with high unmet needs affecting various lysosomal enzymes in the brain. On top of the recent initiation of our clinical trial for other lysosomal storage indication in Batten disease, we will make every effort to bring the clinically effective drugs for children suffering from these life-threatening diseases as soon as possible."
Polaryx Therapeutics, Inc.
Polaryx Therapeutics, Inc. is developing drug candidates for lysosomal storage disorders, for which there are currently no safe and patient-friendly treatment options available. Lysosomal storage disorders are a group of rare inherited genetic disorders caused by the dysfunction of lysosomal enzymes and/or molecules important in the function of these enzymes. Young children are victims of these devastating diseases and die at an early age due to a lack of treatment options.
PLX-300
PLX-300 is a novel, small molecule found in many plants as a deaminated product of phenylalanine. It is widely used as a spice or flavoring material for food. It activates PPARα, which enhances the production of transcription factor EB (TFEB). TFEB then binds to the promoter of genes involved in lysosome biogenesis and activates their production. PLX-300 also has additional activities, such as reducing inflammation and preventing cell death (apoptosis).
Niemann Pick Disease (NPD) types A and B
Niemann Pick Disease types A and B, also known as Acid Sphingomyelinase Deficiency, is caused by genetic mutations in acidic sphingomyelinase (ASMase), the lysosomal enzyme that degrades sphingomyelin. Deficiencies in ASMase or its activity result in a reduced break-down and increased levels of intracellular sphingomyelin leading to cellular dysfunction and cell death. Over time, the cell loss in the organs of children with NPD impairs the function of the brain, lungs, spleen, muscle, and liver with a wide range of symptoms that vary in severity. Especially, sphingomyelin accumulation disrupts the membranes of various subcellular organelles within neurons and glial cells, leading to anomalies in signaling, polarization, calcium homeostasis, synaptic plasticity, and myelin production. These pathologies lead to motor and muscle dysfunction, hearing and vision loss, and early death.





.jpeg)







.jpeg)





.jpg)
.jpg)
.jpg)